II. Síndromes de base genética asociados a autismo
J. Artigas-Pallarés a, E. Gabau-Vila b, M. Guitart-Feliubadaló c SYNDROMIC AUTISM: II. GENETIC SYNDROMES ASSOCIATED WITH AUTISMSummary. Introduction and development. In this study we report on the different genetic syndromes in which autism has been described as one of the possible manifestations. Conclusions. Certain genetic syndromes are providing us with extremely valuable information about the role played by genetics in autism. This is the case of the following syndromes: Angelman syndrome, Prader-Willi syndrome, 15q11-q13 duplication, fragile X syndrome, fragile X premutation, deletion of chromosome 2q, XYY syndrome, Smith-Lemli-Opitz syndrome, Apert syndrome, mutations in the ARX gene, De Lange syndrome, Smith- Magenis syndrome, Williams syndrome, Rett syndrome, Noonan syndrome, Down syndrome, velo-cardio-facial syndrome, myotonic dystrophy, Steinert disease, tuberous sclerosis, Duchenne’s disease, Timothy syndrome, 10p terminal deletion, Cowden syndrome, 45,X/46,XY mosaicism, Myhre syndrome, Sotos syndrome, Cohen syndrome, Goldenhar syndrome, Joubert syndrome, Lujan-Fryns syndrome, Moebius syndrome, hypomelanosis of Ito, neurofibromatosis type 1, CHARGE syndrome and HEADD syndrome. [REV NEUROL 2005; 40 (Supl 1): S151-62] Key words. Angelman syndrome. Autism. Behavioural phenotypes. Fragile X syndrome. Prader-Willi syndrome. DELECIONES Y DUPLICACIONES 15q11-q13
los efectos prácticos son similares a los de la deleción de proce-
En los últimos años ha despertado gran interés en el estudio del
dencia paterna. La mutación de la impronta paterna también
autismo la región 15q11-q13. Se supone que esta región está
genera dos alelos con impronta materna, por tanto, en este caso,
implicada en el 1-4% de los casos de autismo [1,2], con lo cual
ocurre lo mismo que en la deleción paterna (Fig. 1).
se convierte en una de las causas genéticas identificadas más
La figura 2 muestra la región 15q11-q13, distinguiendo la
región correspondiente a la expresión paterna, responsable del
En esta región se pueden distinguir tres situaciones distintas
SPW y la región materna responsable del SA. En un individuo
asociadas a autismo: síndrome de Prader-Willi (SPW) con au-
normal, cada alelo funciona de forma distinta permitiendo única-
tismo, síndrome de Angelman (SA) con autismo y duplicación
mente la expresión paterna o materna en función de la impronta.
El SPW se caracteriza por: hipotonía, hipogonadismo, retraso
mental y obesidad. Su fenotipo conductual, muy característico,
Síndrome de Prader-Willi
consiste en: conducta centrada en la obtención de comida, tozu-
El SPW se genera por una falta de expresión de 15q11-q13 de
dez, conducta manipuladora y síntomas obsesivocompulsivos [3].
origen paterno. Hay que decir que la expresión de 15q11-q13 es
En cuanto al perfil cognitivo, existen buenas capacidades percep-
distinta en el alelo paterno y en el alelo materno; por ello, según
tivas visuoespaciales, si bien la memoria a corto plazo, visual,
deje de expresarse uno u otro, aparecerá el SPW (falta de expre-
motora y auditiva, suele ser débil. Acostumbran a tener buenas
sión de 15q11-q13 de origen paterno) o el SA (falta de expresión
aptitudes domésticas, tales como cocinar y hacer la limpieza. Sin
de 15q11-q13 de origen materno). En el SPW, la desactivación
embargo, sus habilidades sociales son escasas. En general son
de los genes de esta zona puede tener tres orígenes distintos: (1)
muy habladores, con tendencia a ser perseverativos y llevar la
mutación del gen de origen paterno, (2) disomía uniparental
conversación a un abanico de temas, respecto a los cuales son rei-
(DUP) de origen materno, y (3) mutación de la impronta. Esta
terativos. En un estudio que realizamos en 27 pacientes afectos de
región se regula de acuerdo con el mecanismo de la impronta.
SPW mediante el cuestionario de Achenbach; pudimos apreciar
La impronta es el mecanismo (metilación) por el cual ciertos
que algunas de las conductas que manifiestan los niños con SPW
genes, o grupos de genes, son modificados de forma distinta
son habituales en el retraso mental; pero una parte de ellas son
según sean heredados del padre o de la madre, por ello se expre-
específicas del síndrome, es decir no vinculadas al retraso mental.
sa de forma distinta el alelo paterno o materno. Por lo tanto, si
Comparando las respuestas al cuestionario de Achenbach del gru-
existe una deleción en el alelo procedente del padre, la zona
po de pacientes con el SPW con las obtenidas en una muestra de
borrada, no puede ser compensada por el alelo materno a causa
pacientes con retraso mental, de la misma edad y nivel intelec-
tual, las conductas más frecuentes en el grupo SPW y, por tanto,
Si existe una disomía uniparental de origen materno, ambos
supuestamente no ligadas al retraso mental fueron: come dema-
alelos llevan la impronta materna, por este motivo, no puede
siado, roba en casa, le gusta estar solo, roba fuera de casa y se
expresarse la zona que debería aportar el padre. En este caso,
preocupa demasiado por el orden y la limpieza [4].
Por lo menos una parte del fenotipo conductual del SPW,
podría explicarse por una alteración en el metabolismo de la oxi-
Unidad de Neuropediatría. b Unidad de Genética. c Laboratorio de Genéti-ca (UDIAT-Centre Diagnòstic). Hospital de Sabadell. Corporació Sanitària
tocina. Se conoce que la región crítica del SPW del cromosoma
Parc Taulí. Sabadell, Barcelona, España.
15 podría relacionarse con algunas formas de trastorno obsesivo-
Correspondencia: Dr. Josep Artigas. Apartado de Correos 379. E-08200
compulsivo [5]. Se ha visto que los pacientes con trastorno obse-
Sabadell (Barcelona). E-mail: [email protected]
sivo-compulsivo tienen niveles más altos de oxitocina que la
población normal [6]. A su vez, la oxitocina se relaciona con
diversas conductas, entre ellas: la limpieza obsesiva, la agresivi-dad, la regulación del apetito y el apego. Se ha visto que en elSPW están disminuidas las neuronas secretoras de oxitocina enel núcleo paraventricular del hipotálamo. También se ha podidocomprobar que en el SPW existe un elevado nivel de oxitocinaen el líquido cefalorraquídeo. De ello se concluye que los nivelesalterados de oxitocina pueden relacionarse con falta de saciedad,compulsión, pellizcamiento de la piel y otras conductas dentrodel espectro de manifestaciones obsesivocompulsivas [7].
En otro estudio, utilizando el cuestionario Brief, que valora
las funciones ejecutivas a partir de las conductas observadas porlos padres, se identifica un fuerte componente disejecutivo en lamuestra de niños con SPW [8], dato que supone una aproxima-ción al autismo, puesto que la disfunción ejecutiva se sitúa en el
Figura 1. Alteraciones genéticas del síndrome de Prader-Willi.
Si bien la mayor parte de pacientes con SPW no son autis-
tas; se deben tomar en consideración dos hechos. Por un lado
la E6AP-3A ubiquitinproteinligasa, importante para la degrada-
han sido descritos casos que comparten los dos diagnósticos:
ción de proteínas en las neuronas. En los pacientes con SA, el
autismo y SPW [10,11]. Resulta sumamente interesante el he-
retraso en el desarrollo es evidente de forma precoz. La mayoría
cho de que los casos de autismo y SPW coincidan todos con una
de niños con SA pronuncian apenas algunas palabras simples. A
DUP materna, lo cual sugiere que el autismo podría estar vincu-
partir del primer año suelen mostrarse cariñosos, con risa fácil y
lado a una sobreexpresión de un gen ‘materno’ en esta región
apariencia feliz, sin la necesidad de un estímulo apropiado. Las
alteraciones neurológicas también son muy típicas. Hay hipoto-
Por otro lado, analizando detenidamente como se manejan
nía de tronco con hipertonía de extremidades, tendencia a cami-
socialmente y teniendo en cuenta sus disfunciones ejecutivas,
nar con las piernas rígidas, amplia base de sustentación y brazos
emergen una serie de características que permiten aproximar los
flexionados. La falta de equilibrio es habitual. Suele haber tem-
pacientes con SPW a los trastornos del espectro autista (TEA).
blor. Suelen ser hiperactivos, fácilmente excitables, y es caracte-
rístico el movimiento de aleteo de las manos. Es común el estra-
– Dificultad para entender las emociones: las personas con
bismo. También resulta frecuente la existencia de convulsiones,
SPW solo reconocen dos o tres emociones dentro de un con-
a veces de muy difícil control. El perímetro craneal suele estan-
tinuo de felicidad extrema a gran tristeza.
carse. Se observa protusión lingual, babeo y movimientos masti-
– Dificultad para predecir la conducta o estado emocional de
catorios. Los niños con SA acostumbran a sentir fascinación por
el agua y por la música. A veces, la conducta puede parecer agre-
– Problemas para inferir intenciones en los otros.
siva por su tendencia a tirar de los cabellos, pegar y pellizcar.
– Sentido de la justicia, extraordinariamente desarrollado, pe-
Los niños con SA suelen ser fáciles de reconocer, tanto por sus
ro generalmente sesgado en su provecho.
características físicas como conductuales. Sin embargo, dado su
– Egocentrismo, con poca habilidad para monitorizar las con-
retraso profundo, es difícil identificar el autismo en los niños con
SA, sobretodo teniendo en cuenta que algunos síntomas de SA
– Excesiva rigidez e incapacidad para alterar su conducta.
también son propios del autismo; como son: ausencia de lengua-je, aleteo de manos y problemas de relación social. La relación
Estas características, propias del autismo, se han puesto en evi-
genética entre el SA y el autismo consiste en que en casos de
dencia en las personas con SPW mediante la utilización de las
autismo familiar se ha encontrado un desequilibrio en el extremo
siguientes escalas: Autism Behaviour Rating Scale, William
de gen UBE3A, tal como se pudo comprobar en 94 hermanos de
Autism Rating Scale, Australian Asperger Scale y Social Skills
familias con autismo idiopático [14]. Parece pues que este locus
es importante por su efecto aditivo con otros genes relacionados
El interés del SPW con respecto al autismo, no viene tanto a
con el autismo. En un estudio que ha valorado la prevalencia de
partir de la poco frecuente asociación entre ambos trastornos,
autismo en el SA, se encontró que el 42% de pacientes con SA
sino porque permite entender alguno de los mecanismos genéti-
cumplían los criterios de autismo de acuerdo con el Autism
cos y neuroendocrinos implicados en el autismo. Diagnostic Observation Schedule (ADOS). Además los pacien-tes con SA que fueron diagnosticados de autismo con el ADOS
Síndrome de Angelman
también cumplieron las condiciones exigidas por el Autism
El SA tiene una gran similitud genética con el SPW, aunque
Diagnostic Interview-Revised (ADI-R); pero además mostraban
ambos síndromes son clínicamente muy distintos. Las tres alte-
unas puntuaciones más bajas en lenguaje, conducta de adapta-
raciones genéticas descritas para el SPW pueden generar el SA,
ción, y cognición. Estos datos ponen en evidencia la superposi-
pero en este caso la alteración ocurre en el alelo de procedencia
ción que existe entre autismo y SA, y por tanto da soporte a que
materna. Además en el SA también es posible la existencia de
una disregulación en el gen UBE3A desempeña un papel impor-
una mutación en un solo gen, el UBE3A/E6AP [13]. Esta muta-
tante en la base genética del autismo [15].
ción puntual se detecta en el 20-25% de pacientes. Independien-temente de que la causa del SA sea la mutación UBE3A/E6AP, o
Inversión-duplicación 15q11-q13
cualquiera las alteraciones genéticas descritas previamente, el
Las duplicaciones proximales 15q han sido halladas en indivi-
gen responsable del fenotipo del SA es el UBE3A, que codifica
duos con autismo y con diversos grados de retraso mental. En
SÍNDROME X FRÁGIL
Entre el 4 y el 6% de los pacientes au-tistas tiene el síndrome X frágil (SXF)[21-23]. Casi todos los pacientes conel diagnóstico de SXF tienen sínto-mas de autismo (aleteo de manos, malcontacto visual, defensa táctil, len-guaje perseverante y problemas de re-lación social) [24]. Pero, si bien algu-nos síntomas del SXF se pueden con-siderar próximos a los TEA, otras ma-nifestaciones, igualmente típicas del
Figura 2. Región 15q11-13 del cromosoma 15.
SXF, van en sentido contrario. Este esel caso del lenguaje receptivo y de lacapacidad de imitación, que pueden
algunos casos, madres no afectadas presentan la misma duplica-
considerarse puntos relaticamente fuertes del SXF.
ción que sus hijos afectados. En un estudio en el que una madre
La mayoría de estudios sobre prevalencia de autismo en
era portadora de la duplicación, dos hijos autistas habían here-
SXF dan cifras entre el 25-33% [25-27]. El estudio de Sally
dado de su madre la duplicación 15q11-q13, mientras que un
Rogers, que obtiene una prevalencia del 33% de autismo, en
tercer hijo no afectado, no había heredado la duplicación. La
preescolares con SXF, muestra que los niños que tenían ambas
duplicación 15q11-q13 de la madre procedía de novo del cro-
condiciones, mostraban un CI inferior al de los que únicamente
mosoma 15 paterno. Este caso pone en evidencia la importancia
tenían SXF o autismo. Además se ha comprobado que no hay
del origen paterno o materno de la duplicación, puesto que una
correlación entre autismo y número de repeticiones CGG, canti-
herencia paterna de la duplicación produce un fenotipo normal,
dad de FMRP o cantidad de ARNm. Ello sugiere que los niños
en tanto que la herencia materna genera autismo [16]. La expli-
con autismo y SXF tienen un factor genético o ambiental adi-
cación plausible de la razón del distinto impacto de la duplica-
cional que contribuye al desarrollo del autismo. En algunos
ción de procedencia paterna o materna ha de basarse en el fenó-
casos, el factor adicional es obvio, como ocurre cuando el SXF
se asocia a otro síndrome neurológico, pues en estos casos la
Este genotipo se ha encontrado en el 2-4% de autistas de cau-
probabilidad de aparición de autismo se incrementa mucho.
sa desconocida. El fenotipo de estos pacientes comporta fisuras
Esta situación ocurre cuando el SXF se asocia a síndrome de
palpebrales inclinadas hacia abajo, epicantus, raíz nasal amplia,
Down, parálisis cerebral, convulsiones y fenotipo Prader-Willi.
hipotonía, articulaciones metacarpofalángicas hiperextensibles,
En un estudio más reciente, utilizando el ADOS en niños
sindactilia parcial, criptorquidia y en algún caso macrocefalia.
con mutación completa, la prevalencia de autismo y TEA se
Tienen retraso mental o inteligencia límite y es frecuente el tras-
estimó en el 49% (33 y 16%, respectivamente) [28]. Con el uso
torno de déficit de atención/hiperactividad (TDA/H) [17]. No to-
de escalas más inespecíficas como el Developmental Beha-
dos los individuos con duplicación 15q de origen materno, tienen
viour Checklist (DBC-P), se ha obtenido que el 73% de pacien-
TEA. En un estudio con 17 pacientes procedentes de 6 familias,
tes con SXF presentan una puntuación superior al valor de cor-
solo 4 tenían TEA, aunque el resto mostraban déficit sociales o de
te para sospecha de autismo [29]. Ello sugiere que los niños
lenguaje de carácter leve. En dos casos había epilepsia, 11 tenían
con SXF tienden a presentar puntuaciones altas en los instru-
retraso mental leve, 3 tenían capacidad intelectual límite y 3 CI
mentos diagnósticos de autismo, obviamente condicionado por
normal. La mayoría de estos pacientes no tenían rasgos dismórfi-
el hecho de que, al igual que sucede con el SA, ambas condi-
cos, aunque varios mostraban hipotonía, laxitud articular, desvia-
ciones comparten síntomas. Por esta razón, las pruebas de au-
ción de las fisuras palpebrales hacia abajo y labios gruesos [18].
tismo, quizás deberían tener escalas específicas para los pa-
En cierto modo, la duplicación 15q11-q13 actúa de la mis-
ma manera que la DUP de origen materno, capaz de generar
El SXF es la causa genética más frecuentemente hallada en
autismo, tal como se ha comentado en el apartado referente al
el autismo. Por este motivo, la Sociedad Americana de Genética
SPW. La región duplicada puede contener otros genes conti-
Humana, la Academia Americana de Pediatría, la Academia
guos, no vinculados a la impronta (marcador supernumerario
Americana de Neurología [30], y en nuestro país el Grupo de
invertido duplicado del cromosoma 15). Ello hace sospechar la
Estudio de los TEA del Instituto de Salud Carlos III [31], reco-
intervención de estos genes, además del gen UBE3A, como se
miendan el estudio molecular del SXF como rutina de estudio
ha señalado en el SA. Estos genes, tal como se puede ver en la
diagnóstico en los pacientes autistas.
figura 2, son los genes receptores del GABA (ácido gamma-aminobutírico). La función de estos genes aporta interesantessugerencias sobre la genética del autismo, puesto que intervie-
PREMUTACIÓN SXF
nen en la transmisión gabérgica, en el establecimiento de la
Hasta fechas recientes se ha venido considerando que el estado
conectividad neuronal y tienen un papel central en el manteni-
de premutación era totalmente asintomático [32-35]. Sin em-
miento del de tono inhibitorio en el cerebro adulto [19]. Por otro
bargo, progresivamente, se han ido aportando datos en el senti-
lado los agonistas de receptores GABA se han usado para tratar
do contrario. Si bien las manifestaciones clínicas en los indivi-
problemas relacionado con el fenotipo autista, incluyendo las
duos premutados pueden ser sutiles, vale la pena tomarlas en
convulsiones, la ansiedad y la fobia social. El gen candidato de
consideración, pues permiten entender a fondo la enfermedad.
esta región es el gen GABRB3 [20].
La repercusión clínica en la premutación se expresa a nivel físi-
co, cognitivo, emocional, endocrinológico y en relación con un
hermanos de pacientes con SXF el 10% también se podían in-
síndrome de deterioro progresivo en edad avanzada. Por lo que
cluir dentro de los TEA, de acuerdo con el mismo cuestionario
respecta al fenotipo físico, aun sin tener la mutación completa,
[50]. Esto indica que la relación entre autismo y premutación no
se pueden tener rasgos menos aparentes pero dentro del espec-
se puede explicar simplemente por un sesgo de selección de
tro de rasgos físicos propios del SXF [36,37]. En el aspecto
premutados que consultan precisamente por tener algún proble-
cognitivo, hasta fechas recientes, no se habían encontrado datos
ma. Aunque no se conoce la causa de la coincidencia de TEA y
que pudieran sugerir que los individuos premutados estuvieran
premutación, se especula que podría estar en relación con el
afectados. Sin embargo, cuando se han buscado correlaciones
aumento del ARNm, el cual que pudiera tener un efecto tóxico
entre variables cognitivas y nivel de FMRP se han empezado a
sobre las neuronas y los astrocitos [50].
encontrar argumentos positivos en este sentido [38-40]. Al estu-diar aspectos más específicos del aprendizaje en mujeres pre-mutadas se ha hallado un rendimiento más bajo en el cálculo
SÍNDROME DE WILLIAMS
matemático con relación a la lectura y la escritura. Este perfil,
Se caracteriza por la presencia de rasgos físicos peculiares que
hallado en las premutadas, es el mismo, pero menos acentuado,
incluyen: cara de duende, problemas cardíacos y vasculares,
que el de las mujeres con la mutación completa [41]. Así mis-
irritabilidad, anomalías renales y dentales, hiperaucusia y pro-
mo, cuando se ha dirigido el estudio a aspectos neuropsicológi-
blemas músculo esqueléticos. El síndrome de Williams (SW)
cos específicos, tales como la función ejecutiva, también se ha
está causado por una microdeleción del gen de la elastina y del
hallado correlación entre la disfunción ejecutiva y el estado de
gen de un enzima denominado LIM-cinasa [51]. Ambos genes
se ubican en la pequeña zona 7q11.23 [52]. Los niños con SW
El hallazgo clínico más reciente entre los premutados es la
pueden tener prolapso rectal, hernias inguinales e hipercalce-
descripción de un síndrome de temblor cerebeloso y atrofia corti-
mia idiopática, que se manifiesta durante los dos primeros años
cal cerebelosa (FXTAS) en hombres de edad avanzada con la pre-
de vida. Presentan un retraso en su desarrollo motor y cogniti-
mutación FMR1. Nueve abuelos con la premutación y ARNm
vo, sin embargo son muy charlatanes con un discurso muy típi-
elevado desarrollaron temblor cerebeloso y atrofia cortical. Pre-
co por sus deficiencias pragmáticas al cual se le ha denomina-
viamente habían mostrado dificultades para vestirse, conducir,
do cocktail party. También es común que presenten hipersensi-
escribir y comer. La resonancia magnética cerebral mostró: ven-
bilidad a determinados sonidos, tales como los producidos por
trículos dilatados, ligera atrofia cortical y atrofia cerebelosa gra-
aparatos eléctricos, aspiradoras, fuegos artificiales y explosio-
ve. El inicio se sitúa entre los 53-63 años. Las pruebas neuropsi-
nes de globos. A pesar de que pueden destacar sus habilidades
cológicas mostraron pérdida de memoria, además de déficit de
en áreas como leguaje, música y relaciones interpersonales, su
funciones ejecutivas y habilidades visuoespaciales. El curso es
cociente intelectual es bajo (entre 40-85) y tienen serias difi-
progresivo. La conclusión es que un déficit moderado de FMRP o
cultades visuoespaciales [53]. La notable habilidad musical y
un excesivo aumento de FMR1-ARNm pueden generar patología
habilidad verbal de los individuos con SW sugiere que los
retardada del sistema nervioso central (SNC) en un limitado
niños con SW han servido de inspiración para cuentos y leyen-
número de pacientes con la premutación, posiblemente interac-
das. Es prácticamente constante la falta de concentración y la
tuando con otras mutaciones que afectan al SNC [43,44].
fácil distractibilidad. La mayoría de niños con SW tienen una
Al margen de los pacientes con FXTAS, ha sido demostrado
relación bastante pobre con sus compañeros, lo cual les hace
que en los varones premutados están aumentados de tamaño, el
buscar la compañía y relación con los adultos. Pueden estar
cerebelo, el complejo amígdala-hipocampo, el caudado, la ínsu-
muy preocupados, incluso obsesionados por objetos, por per-
la, el tálamo izquierdo y el giro pre y poscentral derecho [45],
sonas o por ciertas actividades Tienen una excesiva ansiedad
tal como se ha visto mediante estudios volumétricos de reso-
por los problemas de salud y por lo que va a ocurrir. Son muy
nancia magnética cerebral. Además, también se ha demostrado
lábiles emocionalmente. Las familias suelen describirlos como
que los cambios en el caudado y la ínsula están relacionados
amigables, cariñosos y pendientes de los sentimientos de los
La relación entre el autismo y la premutación SXF ha adqui-
Si bien los niños con SW en general no pueden ser conside-
rido en los últimos años considerable interés pues nos va a ayu-
rados como autistas, presentan algunas características en su
dar a desvelar alguno de los eslabones en la comprensión del
fenotipo conductual que son propias del autismo, entre las cua-
autismo. Si bien la mayor parte de varones premutados no pre-
les destacan las deficiencias pragmáticas en el lenguaje. La
sentan ninguna manifestación neurocognitiva se ha encontrado
baja capacidad de percepción visual de la globalidad que tie-
que algunos de ellos cumplen los criterios de TEA. La prevalen-
nen los pacientes con SW, también contribuye a disociarlos del
cia de TEA en premutación SXF se ha estimado en 1 de 6 pre-
entorno. Ello les genera dificultad para captar las claves socia-
mutados [47], 4 de 10 [48] y 2 de 7 [49], si bien las muestras de
les transmitidas a partir de lenguaje no verbal. También han
estos estudios son muy sesgadas, por proceder de grupos de pre-
sido descritos pacientes con SW que con cuadros típicos de au-
mutados, reclutados precisamente por tener algún problema. En
un estudio más reciente, cuyos resultados son preliminares, seincluyen 25 pacientes premutados, de los cuales 15 fueron iden-tificados a partir de haber consultado por algún problema neu-
SÍNDROME DE RETT
rocognitivo y 10 fueron seleccionados simplemente por ser her-
El síndrome de Rett (SR) es un trastorno muy severo del des-
manos premutados de pacientes con SXF. En el grupo de pre-
arrollo del sistema nervioso que afecta a las mujeres. Fue des-
mutados que consultaban por algún problema, el 60% tenían
crito por Andreas Rett en 1966 [56]. Los aspectos clínicos más
puntuaciones dentro del rango de TEA en el SCQ (Social Com-
representativos son: regresión psicomotora, movimientos este-
munication Questionnaire); en tanto que entre los premutados
reotipados, marcha atáxica y conducta autística.
Es el único síndrome específico incluido en el DSM-IV
SÍNDROME XYY
entre los trastornos generalizados del desarrollo, con lo cual ya
El síndrome XYY es una trisomía relativamente frecuente, que
se nos da a entender que se trata de una entidad bien definida y
se presenta en uno de cada 894 recién nacidos de sexo masculi-
con unos límites bastante nítidos en comparación con lo que lla-
no. No tienen rasgos dismórficos y su CI, suele estar dentro de
los límites de la normalidad. Sin embargo tienen talla elevada,
Su incidencia se estima entre 1 por 10.000 y 1 por 20.000
trastornos del aprendizaje y problemas emocionales. El desarro-
recién nacidos [57-59]. El desarrollo es aparentemente normal
hasta la edad de 6 a 18 meses. Una vez se inicia la regresión,
En aproximadamente la mitad de los individuos con el sín-
suele ser bastante rápida. Se pierden las habilidades motoras,
drome XYY se describen problemas psicosociales. A partir de
tanto en lo que se refiere al equilibrio y coordinación, como en
la escuela primaria presentan un riesgo mayor que la población
los movimientos propositivos de las manos. Suele aparecer un
normal de tener problemas psiquiátricos, y entre ellos el autis-
leve temblor de las manos y sobre todo movimientos estereoti-
mo, por lo cual muchos de ellos han de seguir programas de
pados consistentes en activad manual de lavado de manos.
educación especial, a pesar de tener inteligencia normal [62,63].
También es posible una tendencia estereotipada a llevarse lasmanos a la boca. Se pierden, si es que se habían adquirido, lashabilidades lingüísticas y la capacidad de comunicación tanto
SÍNDROME DE SMITH-LEMLI-OPITZ
verbal como no verbal. En conjunto su conducta puede definir-
Es un síndrome polimalformativo que comporta retraso mental,
se como autística, por su falta de interacción social, su incapa-
microcefalia, retraso de crecimiento, anomalías craneofaciales,
cidad para comunicarse y la presencia de estereotipias. Muchas
nariz corta con narinas antevertidas, hipogenitalismo, alteracio-
niñas afectas muestran también problemas conductuales y
nes de las extremidades, hipotonía y varias alteraciones viscera-
emocionales, incluyendo ansiedad, bajo estado anímico y con-
les. Este síndrome está causado por una alteración en la biosín-
tesis de colesterol por déficit de 7-dehidrocolesterol reductasa,
Las niñas con SR desarrollan microcefalia y pueden tener
por lo cual podría incluirse entre los errores congénitos del
convulsiones que aparecen entre los 2-4 años. Además es bas-
metabolismo, aunque tradicionalmente se clasifica entre los sín-
tante común la presentación de disfunción respiratoria periódi-
dromes polimalformativos con retraso mental. El gen implicado
ca, trastornos de alimentación, retardo del crecimiento, consti-
se ubica en 11q12-q13. Debe sospecharse este síndrome en los
pacientes en los cuales el autismo se asocia a retraso mental,
El período de regresión rápida es seguido de un plateau con
talla corta y microcefalia [64]. En un estudio con 17 pacientes,
persistencia de las conductas autísticas y el trastorno motor.
se encontró que 9 de ellos, o sea el 53%, cumplían los criterios
El gen causante del SR, MeCP2, fue descubierto en el año
de autismo de acuerdo con el algoritmo del ADI-R [65].
1999 [60]. Se ubica en el brazo largo del cromosoma X (Xq28). Las mujeres tienen dos cromosomas X, por lo cual pareceríadeberían estar menos afectadas. Una explicación posible puede
SÍNDROME DE APERT
ser que la ausencia de una copia funcional del MeCP2 sea letal
Se trata de una mutación autosómica dominante que se caracte-
para el feto masculino antes del nacimiento. Otra cuestión es
riza por acrocefalosindactilia y retraso mental. Pueden estar
porque las mujeres están afectadas, a pesar de que uno de sus
asociadas malformaciones cerebrales como: hidrocefalia, age-
cromosomas X es normal. Esto se debe a la inactivación del cro-
nesia de cuerpo calloso, agenesia de septo, displasia septoópti-
mosoma X, un proceso normal según el cual el cromosoma X es
ca, megalencefalia, trastornos de la migración, encefalocele e
inactivado aleatoriamente en cada célula. Está deficiencia par-
hipoplasia de la sustancia blanca. La mayor parte de casos de
cial permite a las mujeres sobrevivir y desarrollarse normal-
síndrome de Apert son esporádicos, aunque ha sido recogido en
mente durante la primera infancia. A pesar de que puede ser
la literatura algún caso de transmisión paterna [66] o materna
heredado, más del 95% de casos, son mutaciones de novo. El
[67]. El gen implicado está situado en 10q26, zona responsable
gen MeCP2 codifica una proteína que inhibe la función de otros
también de otras craneostenosis, como el síndrome de Crouzon.
genes que deben dejar de actuar de forma sincronizada para
Se ha descrito algún caso aislado asociado a autismo [68].
regular el desarrollo del cerebro. La actuación de ciertos genes,fuera del tiempo que les corresponde, genera alteraciones en eldesarrollo del cerebro
MUTACIONES DEL GEN ARX
El gen ARX (Aristaless-related homeobox, X-linked) ha sido
DELECIÓN 2q37.3
identificado en la región Xp22.13. Este gen es interesante por
La deleción 2q37.3 ha sido hallada en algunos pacientes autis-
haberse descrito diversos cuadros clínicos relacionados con mu-
tas [61]. El fenotipo es muy variable. Los síntomas más cons-
taciones del mismo. Se ha implicado el ARX en: síndrome de
tantes son: bajo peso al nacer, retraso en el desarrollo somático
West [69], epilepsia mioclónica con retraso mental y espasticidad
y mental, alteraciones craneofaciales de ojos, orejas y nariz,
[70], lisencefalia ligada al cromosoma X con genitales ambiguos
cuello corto, deformidades de los dedos y defectos de corazón
[71], retraso mental ligado al cromosoma X [72], síndrome de
y pulmones. Algún paciente puede tener características simila-
Proud (convulsiones, microcefalia adquirida y agenesia del cuer-
res al SR, por presentar dispraxia y estereotipias de lavado de
po calloso) [73] y síndrome de Partington (retraso mental ligado
al X, movimientos distónicos, ataxia y convulsiones). En indivi-
El hallazgo de esta deleción asociada al autismo lleva a con-
duos con estos complejos sindrómicos vinculados al gen ARX
siderar el cromosoma 2 como otro de los candidatos al autismo,
han sido descritos cuadros de autismo típico o conductas autísti-
específicamente en los puntos de rotura de la deleción.
cas [74], por lo cual se debe considerar la mutación del ARX
Esta alteración puede ser detectada por técnicas citogenéticas.
cuando el autismo se asocia a alguno de estos cuadros clínicos. SÍNDROME DE DE LANGE
siones pueden ser niños afables y sensibles a las relaciones in-
El síndrome de De Lange (SDL), cuya alteración genética se si-
terpersonales. También ha sido descrita la asociación de autis-
túa en 5p13.1, se caracteriza por retraso de crecimiento, micro-
mo típico y síndrome de Noonan [81,82].
cefalia, sinofiria, anomalías de las extremidades, hipertricosis,manos y pies pequeños y cara típica. Los rasgos dismórficosque con mayor probabilidad pueden sugerir el diagnóstico es la
SÍNDROME DE DOWN
combinación de: cejas características (bien delimitadas y ar-
Los pacientes con síndrome de Down asocian al retraso mental
queadas), filtrum largo y labios delgados. Por el contrario las
características conductuales, en cierto modo opuestas al autis-
anomalías faciales que con mayor probabilidad inducen a diag-
mo, puesto que suelen ser alegres, simpáticos y con tendencia
nósticos erróneos son: la hipertricosis, la sinofiria y las cejas
espontánea a la sociabilidad. No por ello dejan de ser en oca-
pobladas. El fenotipo conductual, además del retraso mental
siones difíciles de manejar a causa de su tozudez. Son obstina-
(que puede oscilar de límite a profundo), se caracteriza por:
dos y caprichosos. Si bien se detectan problemas psicopatoló-
hiperactividad, tendencia a autolesionarse, trastorno del sueño,
gicos que requieren un diagnóstico y una atención especial,
conducta autística, frecuentemente con estereotipias y altera-
suelen presentarse menos problemas que en otras causas de
ción grave del lenguaje [75]. Además del SDL con todas las
retraso mental. Los trastornos observados más frecuentemente
características típicas, han sido descritas formas leves del sín-
son el trastorno de conducta de oposición desafiante, el TDA/H
drome, con lo cual sus límites pueden resultar imprecisos [76].
y la conducta agresiva. En los adultos es relativamente comúnla depresión. La prevalencia de autismo en el síndrome deDown ha sido estimada entre el 5-9% [83-85]. También se ha
SÍNDROME DE SMITH-MAGENIS
destacado el retraso en el diagnóstico de autismo en los niños
Los pacientes con el síndrome de Smith-Magenis (SMG) pre-
con síndrome de Down [86,87]. De todos modos no se debe
sentan un aspecto típico consistente en braquicefalia, raíz nasal
dejar de señalar que la frecuencia con la que se da el autismo
ancha, aplanamiento facial, boca en forma de cupido y altera-
en el síndrome de Down no sólo no excede la que se puede
ciones en la forma y posición de los pabellones auriculares.
hallar en el retraso mental del mismo nivel que el síndrome de
También suelen tener trastorno del sueño, estatura corta y con-
Down, sino que incluso sea posiblemente inferior [88], por lo
vulsiones. Lo más característico del SMG es el patrón conduc-
cual resulta plausible pensar que el fenotipo conductual del
tual en el cual es muy peculiar la presentación de movimientos
síndrome de Down pueda representar un factor protector para
de abrazo espasmódico, casi patognomónicos de este síndrome.
Son muy manifiestos los problemas de integración sensorialque inciden en su perfil conductual, generando frecuentes tan-trums desencadenados por estímulos irrelevantes. Los niños
SÍNDROME VELOCARDIOFACIAL
afectos de este síndrome son muy demandantes de atención, de
El síndrome velocardiofacial (SVCF), ocasionado por una de-
modo que presentan conductas autoagresivas o heteroagresivas
leción de un pequeño segmento del brazo largo del cromosoma
cuando no reciben la atención exclusiva que tienden a exigir.
22 (22q11), es una de las causas de retraso mental de base
Ello les puede llevar a desarrollar automutilaciones. También es
genética más frecuentes. El síntoma más común, que ha dado
muy común el déficit de atención con o sin hiperactividad.
lugar a la denominación de este síndrome, es la insuficiencia
Aceptan mal los cambios de rutina. Sin embargo, a pesar de
velofaríngea [89]. Otros síntomas son rasgos faciales caracte-
estas conductas disruptivas suelen ser educados, predispuestos a
rísticos (nariz en forma de pera, hendidura palpebral estrecha,
mostrarse agradables y simpáticos [77,78]. El defecto genético
hipertelorismo, retrognatia, hendidura del paladar sin labio
del SMG ha sido identificado como una deleción parcial o total
leporino y displasia de las orejas), voz nasal, tendencia a las
infecciones de oído con posible perdida auditiva y microcefaliaA nivel cardiovascular se describen diversas alteraciones entrela cuales las más frecuentes son las cardiopatías congénitas. SÍNDROME DE NOONAN
También se pueden hallar alteraciones esqueléticas y del creci-
El síndrome de Noonan en un trastorno genético cuyo gen se ha
miento tales como: retraso de crecimiento, hiperexentensibili-
identificado en el brazo largo del cromosoma 12 (12q22), con
dad de los dedos, pie equinovaro y escoliosis. En el aparato
expresión y penetrancia variable [80]. Se trata de un síndrome
genitourinario lo más común es la displasia o agenesia renal,
polimalformativo, caracterizado por cara típica (hipertelorismo,
criptorquidia y la hernia umbilical. Los síntomas neurológicos
cejas arqueadas, orejas de implantación baja, filtrum acanalado,
propios del síndrome son: hipotonía, convulsiones, retardo
base nasal amplia), paladar arqueado, exceso de piel en la nuca,
motor y dificultades de alimentación. Las habilidades psico-
línea de cabello en la nuca de implantación baja, talla corta,
motoras son pobres tanto en lo referente a la motricidad global
retraso puberal, criptorquidia, malformaciones esqueléticas y
cardiopatía congénita. La cardiopatía, generalmente estenosis
El fenotipo cognitivo/conductual se caracteriza por un défi-
pulmonar, la presentan el 80%, aunque la mayoría de las veces
cit intelectual que puede oscilar de límite a retraso mental me-
dio-leve. Si bien es característico el retraso en la aparición del
Desde el punto de vista neurocognitivo, pueden presentar
lenguaje, el perfil cognitivo, en muchas ocasiones encaja en el
retraso mental y/o trastornos del aprendizaje, con problemas
perfil del trastorno de aprendizaje no verbal. Muchos pacientes
visuoconstructivos y de coordinación visuomotora. También
con SVCF son discalcúlicos. Se ha encontrado también un défi-
han sido descritas conductas fóbicas, perseveraciones, terque-
cit en las habilidades visuoperceptivas y visuoespaciales. Tie-
dad y dificultad para la relación con los niños de su edad. Sin
nen dificultad en procesar material nuevo y complejo, pobre
embargo su fenotipo conductal es variable puesto que en oca-
atención visual, buena memoria auditiva y relativamente buenas
habilidades lingüísticas [90]. También existe con frecuencia
de West y con la presencia de tuberomas en lóbulos tempora-
TDA/H. En la escala de Achenbach se detectan puntuaciones
les. Sin embargo, la contribución de estos factores es distinta y
elevadas en aislamiento, problemas sociales y problemas de
complementaria según ha sido sugerido en un trabajo de Asano
et al [100]. El antecedente de síndrome de West se ha relacio-
En general suelen tener labilidad emocional. Al llegar la
nado con problemas de comunicación. Las alteraciones del
adolescencia pueden presentar trastorno bipolar. También es
metabolismo de la glucosa y/o triptófano en cerebelo y cauda-
propio del SVCF la asociación a esquizofrenia. Los trabajos
do medidas por PET, se relacionan con déficit en comunica-
más recientes sugieren que el SVCF se asocia en un 30% de los
ción, interacción social y conducta estereotipada. De acuerdo
casos a trastornos psicóticos. Ello abre interesantes especula-
con estos datos, el autismo se presentaría en los niños con CET,
ciones sobre la posibilidad de que la zona o zonas próximas a
merced a la combinación de diversos efectos negativos. La pre-
22q11, a través de alteraciones en la migración neuronal [92],
sencia de síndrome de West y alteraciones en el córtex tempo-
puedan estar genéticamente implicadas en la esquizofrenia
ral tendrían que ver con retraso mental y alteración en la comu-
nicación, mientras que un desequilibrio metabólico subcortical
En el SVCF ha sido hallado todo el espectro de manifes-
tendría que ver con las conductas estereotipadas y la alteración
taciones del autismo, desde síntomas aislados (‘rasgos autistas’)
a cuadros típicos de autismo, pasando por autismo no especifi-cado [95].
ENFERMEDAD DE DUCHENNE
La enfermedad de Duchenne o distrofia muscular progresiva se
DISTROFIA MIOTÓNICA
presenta en la infancia temprana entre los 2 y 6 años. Los sín-
(ENFERMEDAD DE STEINERT)
tomas principales son: debilidad generalizada y pérdida de teji-
La distrofia miotónica es una enfermedad multisistémica, cuya
do muscular, inicialmente en región torácica y extremidades.
alteración genética está relacionada con la repetición del trinu-
Las pantorrillas tienden a crecer. La progresión es lenta, pero
cleótido CTG en el cromosoma 19. Sigue el fenómeno de anti-
con una probabilidad mínima de supervivencia antes de los 30
cipación. La afectación es multisistémica, si bien la repercusión
años. La herencia es recesiva ligada al X (las mujeres son por-
más importante es a nivel muscular. Las personas afectadas
tadoras). El mecanismo por el cual se produce la debilidad
pueden compartir en distinto grado síntomas de debilidad mus-
muscular es la falta de distrofina, proteína ubicada en la mem-
cular y miotonía, que se suelen expresar mediante una cara
brana de la fibra muscular. Sin embargo, la distrofina también
inexpresiva y ptosis palpebral. Los síntomas extramusculares
tiene actividad funcional en el cerebro y concretamente en el
más comunes son: retraso mental, cataratas, colitis ulcerosa,
córtex, en el cerebelo y en el hipocampo. Por este motivo un
calvicie, alteraciones electrocardiográficas, menopausia precoz
30% de los pacientes afectados de enfermedad de Duchenne
y diversas manifestaciones neuropsiquiátricas tales como de-
tiene alguna afectación intelectual debida a la falta de distrofi-
presión, apatía, falta de motivación e hipersomnia.
na. Los problemas cognitivos afectan a tres áreas: atención,
Se han publicado casos de distrofia miotónica asociados a
aprendizaje verbal e interacción emocional. No sorprende por
autismo [96] y síndrome de Asperger [97]. Al margen de que
tanto que haya sido descrita la asociación de enfermedad de
cumplan criterios de TEA, muchos de ellos tienen un compo-
nente autista, cuya apariencia se acentúa debido a su cara inex-presiva. SÍNDROME DE TIMOTHY
El síndrome de Timothy está producido por mutaciones genéti-
COMPLEJO ESCLEROSIS TUBEROSA
cas espontáneas que interfieren con el funcionamiento de los
El complejo esclerosis tuberosa (CET), es una enfermedad he-
canales de calcio a diversos niveles. Se caracteriza por una afec-
reditaria autonómica dominante que puede ser debida a la mu-
tación multisistémica que incluye arritmias letales, membranas
tación de dos genes distintos: el TSC1 (9q34) y el TSC2
interdigitales, cardiopatía congénita, déficit inmunológico,
(16p13.3), que codifican respectivamente dos proteínas, la
hipoglucemias intermitentes, alteraciones cognitivas y autismo.
hamartina y la tuberina. Ambas proteínas tienen un efecto
Está producida por una mutación en el gen CACNA1C, ubicado
supresor tumoral, mecanismo que puede explicar la presencia
en 12p13.3 [103]. Se ha sugerido que a partir del conocimiento
de tumores en órganos diversos: cerebro, piel, corazón y riño-
del defecto en los canales de calcio implicados se puedan cono-
nes. En el cerebro las lesiones del CET no solo incluyen los
cer mejor las bases biológicas del autismo. Los canales de cal-
tumores (tuberomas), sino también anomalías corticales mi-
cio son proteínas que actúan como poros que controlan el flujo
croscópicas, como: microdisgenesias, heterotopias y defectos
de calcio en el interior de la célula.
de laminación. A nivel más profundo se pueden encontrar nódu-los subependimarios y astrocitomas de células gigantes. Lasconsecuencias de dichas lesiones son la epilepsia, el retraso
DELECIÓN TERMINAL 10p
La deleción terminal 10p se ha asociado a un fenotipo similar al
La incidencia de autismo en el CET se ha estimado entre el
síndrome de DiGeorge: hipoparatriroidismo, sordera sensorial y
17-61% [98,99]. Curiosamente, el autismo vinculado al CET
anomalías renales. Ha sido descrito algún caso que asocia a
se presenta con igual frecuencia en niños que en niñas, dato
estas características retraso mental y autismo. El gen implicado,
que contrasta con el claro predominio del autismo para el sexo
el GATA3, regula el desarrollo de las neuronas serotoninérgicas,
masculino en la población general. La asociación de autismo
por lo cual una disfunción serotoninérgica podría ser el meca-
con el CET, ha sido relacionada con la presencia de síndrome
nismo implicado en el autismo de estos pacientes [104]. SÍNDROME DE COWDEN
– Alteraciones oculares: pigmentación retiniana, degeneración
El síndrome de Cowden es un trastorno hereditario cuyo mayor
tapetorretinal, estrabismo, miopía y hemeralopía.
problema es el desarrollo de hamartomas de piel, mucosas (boca
– Alteraciones somáticas: hipotonía, articulaciones laxas, es-
y nariz) y tracto intestinal. Comporta riesgo de cáncer de pulmón,
coliosis y lordosis, manos y pies afilados, y una variedad de
de tiroides y de útero. Otros síntomas son megacefalia, retraso
otras anormalidades de los pies y dedos.
mental y un raro tumor cerebral no canceroso, la enfermedad de
– Características craneales y faciales: microcefalia y macro-
Lhermitte-Duclos. El gen responsable de esta enfermedad es el
cefalia, filtrum corto, puente nasal alto, paladar arqueado,
PTEN, que está ubicado en 10q23.3. Se trata de un gen supresor
boca abierta e incisivos prominentes.
tumoral, es decir un regulador del crecimiento y división celular.
– Problemas de peso y talla: retraso de crecimiento postnatal,
Puesto que se han descrito casos que asocian el síndrome de
obesidad, sobre todo de la edad de 5 a 11 años.
Cowden con autismo, se debe investigar este síndrome si se de-tecta macrocefalia progresiva en un paciente autista [105].
Otras anormalidades físicas, menos frecuentes, se pueden hallaren corazón, tiroides y genitales. Además pueden presentar, dia-betes, convulsiones febriles y epilepsia. MOSAICISMO 45,X/46,XY
El fenotipo conductual de este síndrome ha sido estudiado
Ha sido descrito un amplio espectro fenotípico en personas con
por Howlin, habiendo destacado una elevada presencia de pro-
mosaicismo 45,X/46,XY. Los individuos con esta alteración
blemas sociales, lingüísticos y conductas ritualistas, de tal mo-
cromosómica se pueden dividir en cuatro grupos: mujeres con
do que la mitad de los pacientes pueden presentar todos los cri-
estigmas de síndrome de Turner, disgenesia gonadal mixta,
pseudohermafrodistismo masculino y fenotipo masculino apa-rentemente normal. En algún caso, correspondiente al gruposíndrome de Turner o disgenesia gonadal mixta, se ha descrito
SÍNDROME DE GOLDENHAR
retraso mental y autismo [106]. También ha sido descrita la aso-
El síndrome de Goldenhar, conocido también como displasia
ciación de esta alteración genética con síndrome de Asperger y
oculoauriculovertebral, es un trastorno polimalformativo, con-
trastorno obsesivocompulsivo, atribuyendo esta asociación a la
génito que comporta malformaciones faciales muy característi-
cas. Los pacientes con este síndrome presentan: pabellón auri-cular incompleto o ausente (microtia), quistes dermoides pre-auriculares, mentón desviado hacia el lado del oído afecto,
SÍNDROME DE MYHRE
comisura bucal que suele estar más elevada en un lado y des-
Se trata de un síndrome polimalformativo en el que se destaca el
arrollo excesivo o ausencia de un ojo. Los niños afectados de
aspecto musculoso, dedos cortos, limitación articular, estatura
síndrome de Goldenhar suelen tener problemas a distintos
corta, sordera y retraso mental. Se añade dismorfia facial (fisu-
niveles: problemas auditivos, debilidad en la motilidad facial
ras palpebrales cortas, hipoplasia maxilar, prognatismo, filtrum
en el lado menos desarrollado, problemas dentales y fusión de
corto, boca pequeña). Se presenta de forma casi exclusiva en
vértebras cervicales. Algunas alteraciones cromosómicas (de-
varones, por lo cual se supone ligado al X.
leción 5q y trisomía 18) se han asociado a síndrome de Golden-
Los individuos afectados suelen tener problemas con la con-
har, pero la causa en la mayoría de alteraciones no se puede de-
ducta social. También ha sido descrita la asociación con cuadros
Aunque el autismo ha sido descrito en pocos casos de sín-
drome de Goldenhar, representa un dato muy interesante, pueslas alteraciones físicas halladas sugieren que la alteración de las
SÍNDROME DE SOTOS
estructuras implicadas ocurre hacia el final del primer trimestre,
Conocido también como gigantismo cerebral. Puede aparecer
lo cual puede hacer pensar que la alteración que genera el autis-
esporádicamente o con una herencia dominante. Se caracteriza
mo se produce probablemente en este periodo [112].
por aceleración del crecimiento somático, con macrocefalia,cara peculiar, frente amplia, paladar arqueado, manos y piesgrandes. Los pacientes afectados suelen ser hipotónicos e hiper-
SÍNDROME DE JOUBERT
laxos, motivo por el cual pueden confundirse con el síndrome X
El síndrome de Joubert es un trastorno autosómico recesivo
frágil. La gran mayoría presentan retraso mental, que puede
caracterizado por hipotonía, hiperpnea/apnea, ataxia truncal,
oscilar de leve a grave, con afectación más marcada en el len-
movimientos oculares anormales y displasia/agenesia de pedún-
guaje. Tienen torpeza motora y problema de coordinación. Su
culos cerebelosos y vermis, y retraso general del desarrollo.
conducta se caracteriza por la presencia de fobias, agresividad,
Otras anomalías menos comunes son movimientos oculares
problemas obsesivos, adherencia a rutinas, trastorno de aten-
anormales, hipersensibilidad al ruido, meningoencefaloceles,
ción y rasgos autistas. La región afectada es 5q35.
microcefalia, orejas de implantación baja, polidactilia, displasia
Han sido descritos casos con cuadros típicos de autismo [109]
retiniana, quistes renales y atresia duodenal.
Se ha asociado el síndrome de Joubert al autismo por la cons-
tatación de que algunos de estos pacientes cumplen todos los cri-terios de autismo. Pero resulta igualmente interesante constatar
SÍNDROME DE COHEN
que los que no pueden ser diagnosticados de autismo, presentan
Se trata de un trastorno genético ligado al cromosoma 8q22, de
al menos alguna de las características nucleares del autismo
herencia autosómica recesiva. Los rasgos físicos que lo caracte-
[113]. La relación entre el síndrome de Joubert y el autismo enfa-
tiza la importancia del cerebelo para entender el autismo. SÍNDROME DE LUJAN-FRYNS NEUROFIBROMATOSIS TIPO 1
El síndrome de Lujan-Fryns forma parte del grupo de retraso men-
La neurofibromatosis tipo 1 (NF1) es el síndrome neurocutáneo
tal ligado al cromosoma X. La peculiaridad de este síndrome vie-
más frecuente. Es un trastorno monogénico de herencia autosó-
ne dada por estar asociado a un hábito marfanoide (retraso mental
mica dominante, de elevada penetrancia, aunque con expresivi-
ligado al X con hábito marfanoide). Los otros datos típicos son la
dad muy variable. Los síntomas cutáneos más típicos son: man-
dismorfia facial y los problemas conductuales. Afecta al sexo mas-
chas café con leche, neurofibromas y nódulos de Lisch. En esta
culino y ha sido asociado a problemas psiquiátricos, entre los que
enfermedad, distintos miembros de la familia, a pesar de tener
destacan la inestabilidad emocional, la hiperactividad, la timidez y
exactamente la misma mutación, gravedad y síntomas, pueden
trastornos psicóticos con alucinaciones visuales y auditivas, por lo
ser muy distintos. Alrededor del 50-70% tiene sólo manchas
cual tiene una fuerte relación con la esquizofrenia [114].
café con leche; en tanto que entre el 30-50% presentan una o
Los casos reportados que muestran la posible asociación
varias manifestaciones importantes, como son el glioma óptico,
con el autismo [115,116] inducen a pensar en este síndrome en
todo autista que presente un fenotipo marfanoide.
Los pacientes autistas tienen un riesgo estimado entre 100 y
190 veces superior al de la población normal de estar afectadosde NF1. Esto sugiere que las dos enfermedades puedan compar-
SÍNDROME DE MOEBIUS
tir una base genética común. Recientemente, el alelo de 6 repe-
El síndrome de Moebius es una diplejía facial congénita que
ticiones AAAT del gen NF1 se observó exclusivamente en los
afecta a los pares craneales VI y VII. Los problemas que se deri-
pacientes autistas graves, pero no en el grupo control. Esto hace
van son: dificultad para la deglución, incapacidad para seguir un
pensar en un papel del gen NF1 en el desarrollo de autismo. Sin
objeto con la mirada sin girar la cabeza, falta de expresión facial
embargo estos datos no han podido ser replicados en otros estu-
e incapacidad para sonreír. La etiología se atribuye a una ausen-
dios, por lo que se recomiendan las investigaciones más exten-
cia congénita de los núcleos motores del VI y VII. Sin embargo,
ello puede ser producido por factores genéticos o ambientales.
Por otro lado, diversos estudios han establecido la frecuen-
Entre estos últimos se han identificado: infecciones, consumo
cia de NF1 en individuos con autismo entre el 0,2 y 14%, con lo
de tóxicos durante la gestación, tales como alcohol, cocaína,
cual se hace difícil discriminar si la asociación responde a una
talidomida y ergotamina o misoprostol (en ocasiones usadas
base etiológica común, o simplemente responde a la probabili-
como drogas abortivas). En algunos casos se ha podido consta-
dad de que un mismo individuo comparta dos trastornos relati-
tar un infarto en los núcleos de pares craneales del tronco cere-
bral, que ha ocurrido durante la vida fetal.
El síndrome de Moebius es muy interesante para el estudio
del autismo, no solo por el elevado número de pacientes que
SÍNDROME CHARGE
asocian ambos trastornos [117], sino porque aportan pistas
CHARGE es el acrónimo que se deriva de los síntomas típicos
sobre el momento del desarrollo en el cual se genera el autismo,
que caracterizan el síndrome: coloboma, malformaciones cardí-
si se tiene en cuenta que los núcleos de los nervios craneales
acas, atresia de coanas, retraso de crecimiento, hipoplasia geni-
implicados se desarrollan en el primer trimestre.
tal, anomalías auriculares y sordera. Se considera que para diag-nosticar el síndrome se requieren por lo menos 4 de los 6 sínto-mas principales. No se conoce la causa y casi siempre es espo-
HIPOMELANOSIS DE ITO
rádico, aunque existe un riesgo de recurrencia estimado en el 1-
Es un síndrome neurocutáneo caracterizado por: áreas de hipo-
2%, puesto que se han encontrado algunos casos familiares.
pigmentación, retraso mental y convulsiones. Las lesiones cutá-
La presencia de retraso metal no es constante. También se
neas pueden estar presentes al nacer, pero también es posible su
ha descrito asociado a autismo [124].
aparición durante los dos primeros años de vida. Adoptan for-mas espirales o en bandas. Las palmas de las manos y de lospies, así como las mucosa suelen estar preservados. Existe una
SÍNDROME HEADD
gran variedad clínica tanto en cuanto a sus manifestaciones,
El síndrome HEADD hace referencia a niños que presentan
como en los hallazgos en la neuroimagen. Además se han des-
hipotonía, epilepsia intratable, autismo, y retraso del desarro-
crito más de 10 alteraciones cromosómicas distintas. Por ello se
llo; y en los que se han hallado alteraciones mitocondriales
duda que sea un síndrome. Independientemente de estas consi-
[125]. El acrónimo que lo define es útil, por sugerir la práctica
deraciones, existen diversas publicaciones que asocian la hipo-
de un estudio mitocondrial en pacientes que reúnan estas ca-
1. Cook EH Jr, Lindgren V, Leventhal BL, Courchesne R, Lincoln A, Shul-
Prader-Willi mediante el cuestionario CBCL 418 de Achenbach. Rev
man C, et al. Autism or atypical autism in maternally but not paternally
derived proximal 15q duplication. Am J Hum Genet 1997; 60: 928-34.
5. Leckman JF, Goodman WK, North WG, Chappell PB, Price LH,
2. Thomas JA, Johnson J, Peterson-Kraai TL, Wilson R, Tartaglia N,
Pauls D, et al. The role of central oxytocin in obsessive compulsive
LeRoux J, et al. Genetic and clinical characterization of patients with
disorder and related normal behavior. Psychoneuroendocrinology
an interstitial duplication 15q11-q13, emphasizing behavioral pheno-
type and response to treatment. Am J Hum Genet 2003; 119: 111-20.
6. Swaab DF, Purba JS, Hofman MA. Alterations in the hypothalamic
3. Artigas-Pallarés J. Fenotipos conductuales. Rev Neurol 2002; 34 (Supl
paraventricular nucleus and its oxytocin neurons (putative satiety cells)
in Prader-Willi syndrome: a study of five cases. J Clin Endocrinol
4. Artigas J, Gabau E. Estudio del fenotipo conductual del síndrome de
7. Swaab DF, Purba JS, Hofman MA. Alterations in the hypothalamic
phenotype of female carriers of fragile X: additional evidence for
paraventricular nucleus and its oxytocin neurons (putative satiety cells)
specificity. J Dev Behav Pediatr 1993; 14: 328-35.
in Prader-Willi syndrome: a study of five cases. J Clin Endocrinol
34. Taylor AK, Safanda JF, Fall MZ, Quince C, Lang KA, Hull CE, et al.
Molecular predictors of cognitive involvement in female carriers of
8. Artigas-Pallares J. Disfunciones cognitivas en el síndrome de Prader-
fragile X syndrome. JAMA 1994; 271: 507-14.
Willi. II Congreso Nacional. Síndrome de Prader-Willi. Barcelona, 22-
35. Mazzocco MM, Holden JJ. Neuropsychological profiles of three sis-
ters homozygous for the fragile X premutation. Am J Med Genet 1996;
9. Griffith EM, Pennington BF, Wehner EA, Rogers SJ. Executive func-
tions in young children with autism. Child Dev 1999; 70: 817-32.
36. Riddle JE, Cheema A, Sobesky WE, Gardner SC, Taylor AK, Penning-
10. Cassidy SB. Morris CA. Behavioral phenotypes in genetic syndromes:
ton BF, et al. Phenotypic involvement in females with the FMR1 gene
genetic clues to human behavior. Adv Pediatr 2002; 49: 59-86.
mutation. Am J Ment Retard 1998; 102: 590-601.
11. Schroer RJ, Phelan MC, Michaelis RC, Crawford EC, Skinner SA,
37. McConkie-Rosell A, Lachiewicz AM, Spiridigliozzi GA, Tarleton J,
Cuccaro M, et al. Autism and maternally derived aberrations of chro-
Schoenwald S, Phelan MC, et al. Evidence that methylation of the
mosome 15q. Am J Hum Genet 1998; 76: 327-36.
FMR-I locus is responsible for variable phenotypic expression of the
12. Whitman BY, Greenswag LR. Psychological and behavioural manage-
fragile X syndrome. Am J Hum Genet 1993; 53: 800-9.
ment. In Greenswag LR, Alexander RC, eds. Management of Prader-
38. Hagerman RJ, Staley LW, O’Conner R, Lugenbeel K, Nelson D, Mc
Willi syndrome. 2 ed. New York; Springer-Verlag; 1995. p. 125-41.
Lean SD, et al. Learning-disabled males with a fragile X CGG expan-
13. Kishino T, Lalande M, Wagstaff J. UBE3A/E6AP mutations cause
sion in the upper premutation size range. Pediatrics 1996; 97: 122-6.
Angelman syndrome. Nat Genet 1997; 15: 70-3.
39. Kaufmann WE, Abrams MT, Chen W, Reiss AL. Genotype, molecular
14. Nurmi EL, Bradford Y, Chen Y, Hall J, Arnone B, Gardiner MB, et al.
phenotype, and cognitive phenotype: correlations in fragile X syn-
Linkage disequilibrium at the Angelman syndrome gene UBE3A in
drome. Am J Med Genet 1999; 83: 286-95.
autism families. Genomics 2001; 77: 105-13.
40. Tassone F, Hagerman RJ, Taylor AK, Mills JB, Harris SW, Gane LW,
15. Peters SU, Beaudet AL, Madduri N, Bacino CA. Autism in Angelman
et al. Clinical involvement and protein expression in individuals with
syndrome: implications for autism research. Clin Genet 2004; 66: 530-6.
the FMR1 premutation. Am J Med Genet 2000; 91: 144-52.
16. Cook EH Jr, Lindgren V, Leventhal BL, Courchesne R, Lincoln A,
41. Lachiewicz AM, Dawson DV, McConkie-Rosell A, Spiridigliozzi GA.
Shulman C, et al. Autism or atypical autism in maternally but not
Mathematics weakness in premutation females with the fragile X syn-
paternally derived proximal 15q duplication. Am J Hum Genet 1997;
drome. 7th International Fragile X Conference. Los Angeles; July 2000.
42. Hills JL, Wilson R, Sobesky W, Harris SW, Grigsby J, Butler E, et al.
17. Thomas JA, Johnson J, Peterson-Kraai TL, Wilson R, Tartaglia N,
Executive functioning deficits in adult males with the fragile X premu-
LeRoux J, et al. Genetic and clinical characterization of patients with
tation: an emerging phenotype. 7th International Fragile X Confer-
an interstitial duplication 15q11-q13, emphasizing behavioral pheno-
type and response to treatment. Am J Hum Genet 2003; 119: 111-20.
43. Hagerman RJ, Tassone F, Leehey M, Hills J, Wilson R, Landau W, et
18. Bolton PF, Dennis NR, Browne CE, Thomas NS, Veltman MW,
al. Cerebellar tremor and cerebellar cortical atrophy in older males
Thompson RJ, et al. The phenotypic manifestations of interstitial
with the fragile X premutation. 7th International Fragile X Conference.
duplications of proximal 15q with special reference to the autistic
spectrum disorders. Am J Hum Genet 2001; 105: 675-85.
44. Hagerman RJ. The physical and behavioral phenotype. In Hagerman RJ,
19. Owens DF, Kriegstein AR. Is there more to GABA than synaptic inhi-
Hagerman PJ, eds. Fragile X syndrome. Diagnosis, treatment and
bition? Nat Rev Neurosci 2002; 3: 715-27.
research. 3 ed. Baltimore: Johns Hopkins University Press; 2002. p. 3-110.
20. Buxbaum JD, Silverman JM, Smith CJ, Greenberg DA, Kilifarski M,
45. Daly ED, Moore CJ, Schmitz N, Jacobs P, Davis K, Murphy KC, et al.
Reichert J, et al Association between a GABRB3 polymorphism and
Premutation expansion of CGG triplet repeats affects brain; a study of
autism. Mol Psychiatry 2002; 7: 311-6.
male carriers of fragile X syndrome. 9th Annual Scientific Meeting. Soci-
21. Brown WT, Jenkins EC, Cohen IL, Fisch GS, Wolf-Schein EG, Gross
ety for the Study of Behavioural Phenotypes. Oxford; November 2001.
A, et al. Fragile X and autism: a multicenter survey. Am J Hum Genet
46. Moore CJ , Daly ED, Tassone F, Jacobs P, Davis K, Murphy KC, et al.
Neuroanatomical effect of FMR1 gene mRNA in premutation carriers
22. Bailey A, Bolton P, Butler L, Le Couteur A, Murphy M, Scott S, et al.
of fragile-X syndrome. 9th Annual Scientific Meeting. Society for the
Prevalence of the fragile X anomaly amongst autistic twins and single-
Study of Behavioural Phenotypes. Oxford; November 2001.
tons. J Child Psychol Psychiatry 1993; 34: 673-88.
47. Tassone F, Hagerman RJ, Taylor AK, Mills JB, Harris SW, Gane LW,
23. Wassink TH, Piven J, Patil SR. Chromosomal abnormalities in a clinic
et al. Clinical involvement and protein expression in individuals with
sample of individuals with autistic disorder. Psychiatr Genet 2001; 11:
the FMR1 premutation. Am J Hum Genet 2000; 91: 144-52.
48. Aziz M, Stathopulu E, Callias M, Taylor C, Turk J, Oostra B, et al.
24. Artigas-Pallarés J, Brun C, Gabau E. Aspectos médicos y neuropsi-
Clinical features of boys with fragile X premutations and intermediate
cológicos del síndrome X frágil. Rev Neurol Clin 2001; 2: 42-54.
alleles. Am J Hum Genet 2003; 121: 119-27.
25. Hagerman RJ, Jackson AW III, Levitas A, Rimland B, Braden M. An
49. Brun C, Artigas J, Ramírez A, Lorente I, Gabau E, Milà M. Manifesta-
analysis of autism in fifty males with the fragile X syndrome. Am J
ciones clínicas de la premutación frágil X en niños. Rev Neurol 2001;
26. Bailey A, Palferman S, Heavey L, Le Couteur A. Autism: the pheno-
50. Farzin F, Perry H, Hessl D, Loesch DZ, Cohen J, Gane LW, et al. Phe-
type in relatives. J Autism Dev Disord 1998; 28: 369-92.
notype of the fragile X premutation in childhood: a preliminary study
27. Rogers SJ, Wehner DE, Hagerman R. The behavioral phenotype in
[poster presentation]. 9th International Fragile X Conference. Wash-
fragile X: symptoms of autism in very young children with fragile X
syndrome, idiopathic autism, and other developmental disorders. J Dev
51. Ewart AK, Morris CA, Atkinson D, Jin W, Sternes K, Spallone P, et al.
Hemizygosity at the elastin locus in a developmental disorder, Wi-
28. Harris SW, Hagerman RJ. ADOS-G profiles in males with fragile X.
lliams syndrome. Nat Genet 1993; 5: 11-6.
9th International Fragile X Conference. Washington, DC; 2004.
52. Frangiskakis JM, Ewart AK, Morris CA, Mervis CB, Bertrand J, Ro-
29. Artigas J, Brun C. Assessment of behavioural phenotype of fragile X
binson BF, et al. LIM-kinase1 hemizygosity implicated in impaired
syndrome with DBC-P. 9th International Fragile X Conference. Wash-
visuospatial constructive cognition. Cell 1996; 86: 59-69.
53. Udwin O, Yule W. A cognitive and behavioural phenotype in Williams
30. Filipek PA, Accardo PJ, Ashwal S, Baranek GT, Cook EH Jr, Dawson
syndrome. J Clin Exp Neuropsychol 1991; 13: 232-44.
G, et al. Practice parameter: screening and diagnosis of autism: report
54. Reiss AL, Feinstein C, Rosenbaum KN, Borengasser-Caruso MA,
of the Quality Standards Subcommittee of the American Academy of
Goldsmith BM. Autism associated with Williams syndrome. J Pediatr
Neurology and the Child Neurology Society. Neurology 2000; 55:
55. Gillberg C, Rasmussen P. Brief report: four case histories and a litera-
31. Guía de buenas prácticas para el proceso diagnóstico en los trastornos
ture review of Williams syndrome and autistic behavior. J Autism Dev
del espectro autista (TEA). Grupo de Estudio de los TEA del Instituto
de Salud Carlos III, Ministerio de Sanidad y Consumo, España. Rev
56. Rett A. Cerebral atrophy associated with hyperammonaemia. In Vin-
ken PJ, Bruyn GW, Klawans HL, eds. Metabolic and deficiency dis-
32. Reiss AL, Freund L, Abrams MT, Boehm C, Kazazian H. Neurobehav-
eases of the nervous system III. Handbook of Clinical Neurology. Vol
ioral effects of the fragile X premutation in adult women: a controlled
26. Amsterdam: Elsevier/North Holland Biomedical Press; 1977.
study. Am J Hum Genet 1993; 52: 884-94.
57. Kerr AM, Stephenson JBP. Rett’s syndrome in the west of Scotland.
33. Mazzocco MM, Pennington BF, Hagerman RJ. The neurocognitive
58. Hagberg B. Rett syndrome: Swedish approach to analysis of preva-
85. Turk J. Children with Down’s syndrome and fragile X syndrome: a
lence and cause. Brain Dev 1985; 7: 277-80.
comparison study. Society for the Study of Behavioural Phenotypes.
59. Burd L, Vesley B, Martsolf JT, Kerbeshian J. Prevalence study of Rett
2nd Symposium Abstracts. Oxford; 1992.
syndrome in North Dakota children. Am J Med Genet 1991; 38: 565-8.
86. Rasmussen P, Borjesson O, Wentz E, Gillberg C. Autistic disorders in
60. Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi
Down syndrome: background factors and clinical correlates. Dev Med
HY. Rett syndrome is caused by mutations in X-linked MECP2, encod-
ing methyl-CpG-binding protein 2. Nat Genet 1999; 23: 185-8.
87. Howlin P, Wing L, Gould J. The recognition of autism in children with
61. Lukusa T, Vermeesch JR, Holvoet M, Fryns JP, Devriendt K. Deletion
Down syndrome -implications for intervention and some speculations
2q37.3 and autism: molecular cytogenetic mapping of the candidate
about pathology. Dev Med Child Neurol 1995; 37: 398-414.
region for autistic disorder. Genet Couns 2004; 15: 293-301.
88. Rasmussen P, Borjesson O, Wentz E, Gillberg C. Autistic disorders in
62. Geerts M, Steyaert J, Fryns JP. The XYY syndrome: a follow-up study
Down syndrome: background factors and clinical correlates. Dev Med
on 38 boys. Genet Couns 2003; 14: 267-79.
63. Weidmer-Mikhail E, Sheldon S, Ghaziuddin M. Chromosomes in
89. Vantrappen G, Rommel N, Devriendt K, Cremers CW, Feenstra L,
autism and related pervasive developmental disorders: a cytogenetic
Fryns JP. Clinical features in 130 patients with the velo-cardio-facial
study. J Intellec Disabil Res 1998; 42: 8-12.
syndrome. The Leuven experience. Acta Otorhinolaryngol Belg 2001;
64. Goldenberg A, Chevy F, Bernard C, Wolf C, Cormier-Daire V. Clinical
characteristics and diagnosis of Smith-Lemli-Opitz syndrome and ten-
90. Swillen A, Vandeputte L, Cracco J, Maes B, Ghesquiere P, Devriendt
tative phenotype-genotype correlation: report of 45 cases. Arch Pediat
K, et al. Neuropsychological, learning and psychosocial profile of pri-
mary school aged children with the velo-cardio-facial syndrome
65. Tierney E, Nwokoro NA, Porter FD, Freund LS, Ghuman JK, Kelley
(22q11 deletion): evidence for a nonverbal learning disability? Child
RI. Behavior phenotype in the RSH/Smith-Lemli-Opitz syndrome. Am
91. Swillen A, Devriendt K, Ghesquiere P, Fryns JP. The behavioural phe-
66. Rollnick BR. Male transmission of Apert syndrome. Clin Genet 1988;
notype in child and adolescents with VCFS (del22q11). 9th Annual
Scientific Meeting. Society for the Study of Behavioural Phenotypes.
67. Roberts KB, Hall JG. Apert’s acrocephalosyndactyly in mother and
daughter: cleft palate in the mother. Birth Defects Orig Art Ser 1971; 7:
92. Bird LM, Scambler P. Cortical dysgenesis in 2 patients with chromo-
some 22q11 deletion. Clin Genet 2000; 58: 64-8.
68. Morey-Canellas J, Sivagamasundari U, Barton H. A case of autism in
93. Jurewicz I, Owen RJ, O’Donovan MC, Owen MJ. Searching for sus-
a child with Apert’s syndrome. Eur Child Adolesc Psychiatry 2003;
ceptibility genes in schizophrenia. Eur Neuropsychopharmacol 2001;
69. Bruyere H, Lewis MES, Wood S, MacLeod PJ, Langlois S. Confirma-
94. Murphy KC, Owen MJ. Velo-cardio-facial syndrome: a model for
tion of linkage in X-linked infantile spasms (West syndrome) and
understanding the genetics and pathogenesis of schizophrenia. Br J
refinement of the disease locus to Xp21.3-Xp22.1. Clin Genet 1999;
95. Niklasson L, Rasmussen P, Oskarsdottir S, Gillberg C. Chromosome
70. Scheffer IE, Wallace RH, Phillips FL, Hewson P, Reardon K, Parasi-
22q11 deletion syndrome (CATCH 22): neuropsychiatric and neu-
vam G, et al. X-linked myoclonic epilepsy with spasticity and intellec-
ropsychological aspects. Dev Med Child Neurol 2002; 44: 44-50.
tual disability: mutation in the homeobox gene ARX. Neurology 2002;
96. Yoshimura I, Sasaki A, Akimoto H, Yoshimura N. A case of congenital
myotonic dystrophy with infantile autism. Brain Dev 1989; 21: 379-84.
71. Bonneau D, Toutain A, Laquerriere A, Marret S, Saugier-Veber P,
97. Paul M, Allington-Smith P. Asperger syndrome associated with Stein-
Barthez MA. X-linked lissencephaly with absent corpus callosum and
ert's myotonic dystrophy. Dev Med Child Neurol 1997; 39: 280-1.
ambiguous genitalia (XLAG): clinical, magnetic resonance imaging,
98. Hunt A, Shepherd C. A prevalence study of autism in tuberous sclero-
and neuropathological findings. Ann Neurol 2002; 51: 340-9.
sis. J Autism Dev Disord 1993; 23: 323-39.
72. Bienvenu T, Poirier K, Friocourt G, Bahi N, Beaumont D, Fauchereau
99.Gillberg IC, Gillberg C, Ahlsen G. Autistic behaviour and attention
F et al. ARX, a novel Prd-class-homeobox gene highly expressed in the
deficits in tuberous sclerosis: a population-based study. Dev Med Child
telencephalon, is mutated in X-linked mental retardation. Hum Mol
100. Asano E, Chugani DC, Muzik O, Behen M, Janisse J, Rothermel R.
73. Proud VK, Levine C, Carpenter NJ. New X-linked syndrome with
Autism in tuberous sclerosis complex is related to both cortical and
seizures, acquired micrencephaly, and agenesis of the corpus callosum.
subcortical dysfunction. Neurology 2001; 57: 1269-77.
101. Zwaigenbaum L, Tarnopolsky M. Two children with muscular dystro-
74. Turner G, Partington M, Kerr B, Mangelsdorf M, Gecz J. Variable
phies ascertained due to referral for diagnosis of autism. J Autism Dev
expression of mental retardation, autism, seizures, and dystonic hand
movements in two families with an identical ARX gene mutation. Am
102. Kumagai T, Miura K, Ohki T, Matsumoto A, Miyazaki S, Nakamura
M, et al. Central nervous system involvements in Duchenne/Becker
75. Ruggieri VL, Arberas CL. Fenotipos conductuales. Patrones neuropsi-
muscular dystrophy. Brain Dev 2001; 33: 480-6.
cológicos biológicamente determinados. Rev Neurol 2003; 37: 239-53.
103. Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et
76. Berney TP, Ireland M, Burn J. Behavioural phenotype of Cornelia de
al. Ca1.2 calcium channel dysfunction causes a multisystem disorder
Lange syndrome. Arch Dis Child 1999; 81: 333-6.
including arrhythmia and autism. Cell 2004; 119: 19-31.
77. Dykens EM, Smith AC. Distinctiveness and correlates of maladaptive
104. Verri A, Maraschio P, Devriendt K, Uggetti C, Spadoni E, Haeusler E,
behaviour in children and adolescents with Smith-Magenis syndrome.
et al. Chromosome 10p deletion in a patient with hypoparathyroidism,
J Intellect Disabil Res 1998; 42: 481-9.
severe mental retardation, autism and basal ganglia calcifications. Ann
78. Blanco-Barca O, Gallego-Blanco M, Ruiz-Ponte C, Barros-Angueira
F, Esquete-López C, Eirís-Puñal J, et al. Síndrome de Smith-Magenis:
105. Goffin A, Hoefsloot LH, Bosgoed E, Swillen A, Fryns JP. PTEN muta-
aportación de dos nuevos casos y aproximación a su característico
tion in a family with Cowden syndrome and autism. Am J Med Genet
fenotipo conductual. Rev Neurol 2004; 38: 1038-42.
79. Park JP, Moeschler JB, Davies WS, Patel PI, Mohandas TK. Smith-
106. Telvi L, Lebbar A, Del Pino O, Barbet JP, Chaussain JL. 45,X/46,XY
Magenis syndrome resulting from a de novo direct insertion of proxi-
mosaicism: report of 27 cases. Pediatrics 1999; 104: 304-8.
mal 17q into 17p11.2. Am J Med Genet 1998; 77: 23-7.
107. Fontenelle LF, Mendlowicz MV, Bezerra de Menezes G, Dos Santos
80. Brady AF, Jamieson CR, Van der Burgt I, Crosby A, Van Reen M, Kremer
Martins RR, Versiani M. Asperger syndrome, obsessive-compulsive
H, et al. Further delineation of the critical region for Noonan syndrome on
disorder, and major depression in a patient with 45,X/46,XY
the long arm of chromosome 12. Eur J Hum Genet 1997; 5: 336-7.
mosaicism. Psychopathology 2004; 37: 105-9.
81. Paul R, Cohen DJ, Volkmar FR. Autistic behaviors in a boy with Noo-
108. Titomanlio L, Marzano MG, Rossi E, D’Armiento M, De Brasi D,
nan syndrome. J Autism Dev Disord 1983; 13: 433-4.
Vega GR, et al. Case of Myhre syndrome with autism and peculiar skin
82. Ghaziuddin M, Bolyard B, Alessi N. Autistic disorder in Noonan syn-
histological findings. Am J Hum Genet 2001; 103: 163-5.
drome. J Intellect Disabil Res 1994; 38: 67-72.
109. Morrow JD, Whitman BY, Accardo PJ. Autistic disorder in Sotos syn-
83. Ghaziuddin M. Autism in Down’s syndrome: family history correlates.
drome: a case report. Eur J Ped 1990; 149: 567-9.
J Intellect Disabil Res 1997; 41: 86-91.
110. Tantam D, Evered C, Hersov L. Asperger's syndrome and ligamentous
84. Kent L, Evans J, Paul M, Sharp M. Comorbidity of autistic spectrum
laxity. J Am Acad Child Adolesc Psychiatry 1990; 29: 892-6.
disorders in children with Down syndrome. Dev Med Child Neurol
111. Howlin P. Autistic features in Cohen syndrome: a preliminary report.
Dev Med Child Neurol 2001; 43: 692-6.
112. Landgren M, Gillberg C, Stromland K. Goldenhar syndrome and autis-
CR, eds. Handbook of neurodevelopmental and genetic disorders in
tic behaviour. Dev Med Child Neurol 1992; 34: 999-1005.
children. New York: Guilford Press; 1999. p. 350-67.
113. Ozonoff S, Williams BJ, Gale S, Miller JN. Autism and autistic behav-
120. Plank SM, Copeland-Yates SA, Sossey-Alaoui K, Bell JM, Schroer RJ,
ior in Joubert syndrome. J Child Neurol 1999; 14: 636-41.
Skinner C, et al. Lack of association of the (AAAT)6 allele of the
114. De Hert M, Steemans D, Theys P, Fryns JP, Peuskens J. Lujan-Fryns
GXAlu tetranucleotide repeat in intron 27b of the NF1 gene with
syndrome in the differential diagnosis of schizophrenia. Am J Med
autism. Am J Hum Genet 2001; 105: 404-5.
121. Mbarek O, Marouillat S, Martineau J, Barthelemy C, Muh JP, Andres
115. Swillen A, Hellemans H, Steyaert J, Fryns JP. Autism and genetics:
C. Association study of the NF1 gene and autistic disorder. Am J Hum
high incidence of specific genetic syndromes in 21 autistic adolescents
and adults living in two residential homes in Belgium. Am J Hum
122. Gillberg C, Forsell C. Childhood psychosis and neurofibromatosis -
more than a coincidence? J Autism Dev Disord 1984; 14: 1-8.
116. Gurrieri F, Neri G. A girl with the Lujan-Fryns syndrome. Am J Hum
123. Mouridsen SE, Andersen LB, Sorensen SA, Rich B, Isager T. Neurofi-
bromatosis in infantile autism and other types of childhood psychoses.
117. Gillberg C, Steffenburg S. Autistic behaviour in Moebius syndrome.
124. Fernell E, Olsson VA, Karlgren-Leitner C, Norlin B, Hagberg B, Gill-
118. Hermida A, Eirís J, Álvarez-Moreno A, Alonso-Martín A, Barreiro J,
berg C. Autistic disorders in children with CHARGE association. Dev
Castro-Gago M. Hipomelanosis de Ito. Un síndrome neurocutáneo
heterogéneo y posiblemente infradiagnosticado. Rev Neurol 1997; 25:
125. Fillano JJ, Goldenthal MJ, Rhodes CH, Marín-García J. Mitochondrial
dysfunction in patients with hypotonia, epilepsy, autism, and develop-
119. Nilson D, Bradford LW. Neurofibromatosis. In Goldstein S, Reynolds
mental delay: HEADD syndrome. J Child Neurol 2002; 17: 435-9. EL AUTISMO SINDRÓMICO: II. SÍNDROMES O AUTISMO SINDRÓMICO: II. SÍNDROMAS DE BASE GENÉTICA ASOCIADOS A AUTISMODE BASE GENÉTICA ASSOCIADOS AO AUTISMOResumen. Introducción y desarrollo. Se describen los distintos sín- Resumo. Introdução e desenvolvimento. Descrevem-se os distin- dromes de base genética, en los cuales ha sido descrito el autismotos síndromas de base genética, nos quais se descreveu o autismocomo una de sus posibles manifestaciones. Conclusiones. Determi-como uma das suas possíveis manifestações. Conclusões. Deter-nados síndromes genéticos están aportando información muyminados síndromas genéticos detêm informação muito valiosavaliosa sobre el conocimiento de la genética del autismo. Este es elsobre o conhecimento da genética do autismo. Este é o caso doscaso de los siguientes síndromes: síndrome de Angelman, síndromeseguintes síndromas: síndroma de Angelman, síndroma de Pra-de Prader-Willi, duplicación 15q11-q13, síndrome X frágil, premu-der-Willi, duplicação 15q11-q13, síndroma X frágil, premutaçãotación del síndrome X frágil, deleción del cromosoma 2q, síndromedo síndroma X frágil, deleção do cromossoma 2q, síndroma XYY,XYY, síndrome de Smith-Lemli-Opitz, síndrome de Apert, mutacio-síndroma de Smith-Lemli-Opitz, síndroma de Apert, mutações dones del gen ARX, síndrome de De Lange, síndrome de Smith-Ma-gene ARX, síndroma de De Lange, síndroma de Smith-Magenis,genis, síndrome de Williams, síndrome de Rett, síndrome de Noo-síndroma de Williams, síndroma de Rett, síndroma de Noonan,nan, síndrome de Down, síndrome velocardiofacial, distrofia mio-síndroma de Down, síndroma velocardiofacial, distrofia miotóni-tónica, enfermedad de Steinert, esclerosis tuberosa, enfermedad deca, doença de Steinert, esclerose tuberosa, doença de Duchenne,Duchenne, síndrome de Timothy, deleción terminal 10p, síndromesíndroma de Timothy, deleção terminal 10p, síndroma de Cow-de Cowden, mosaicismo 45 X/46 XY, síndrome de Myhre, síndromeden, mosaicismo 45 X/46 XY, síndroma de Myhre, síndroma dede Sotos, síndrome de Cohen, síndrome de Goldenhar, síndrome deSotos, síndroma de Cohen, síndroma de Goldenhar, síndroma deJoubert, síndrome de Lujan-Fryns, síndrome de Moebius, hipome-Joubert, síndroma de Lujan-Fryns, síndroma de Moebius, hipo-lanosis de Ito, neurofibromatosis tipo 1, síndrome CHARGE y sín-melanose de Ito, neurofibromatose tipo 1, síndroma CHARGE edrome HEADD. [REV NEUROL 2005; 40 (Supl 1): S151-62]síndroma HEADD. [REV NEUROL 2005; 40 (Supl 1): S151-62]Palabras clave. Autismo. Fenotipos conductuales. Síndrome de An- Palavras chave. Autismo. Fenótipos de conduta. Síndroma de An- gelman. Síndrome de Prader-Willi. Síndrome X frágil.gelman. Síndroma de Prader-Willi. Síndroma X frágil.
Applications of Vibrational Spectroscopy in Criminal Forensic Analysis Edward G. Bartick Handbook of Vibrational Spectroscopy John M. Chalmers and Peter R. Griffiths (Editors) John Wiley & Sons Ltd, Chichester, 2002 Applications of Vibrational Spectroscopy in Criminal Forensic Analysis Edward G. Bartick FBI Academy, Quantico, VA, USA This is publication number 01-06 of
TRIPTANS FOR MIGRAINE The triptans are very effective for the treatment of an acute migraine attack even if taken several hours after the onset of the pain. This feature makes them quite useful for you when you awaken with a fully developed migraine. However, they are most effective if taken at the onset of the headache when the pain is mild. In studies comparing the triptans to placebo, t
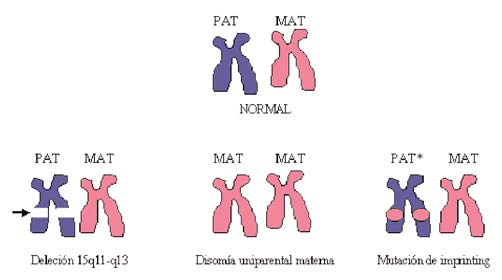 diversas conductas, entre ellas: la limpieza obsesiva, la agresivi-dad, la regulación del apetito y el apego. Se ha visto que en elSPW están disminuidas las neuronas secretoras de oxitocina enel núcleo paraventricular del hipotálamo. También se ha podidocomprobar que en el SPW existe un elevado nivel de oxitocinaen el líquido cefalorraquídeo. De ello se concluye que los nivelesalterados de oxitocina pueden relacionarse con falta de saciedad,compulsión, pellizcamiento de la piel y otras conductas dentrodel espectro de manifestaciones obsesivocompulsivas [7].
diversas conductas, entre ellas: la limpieza obsesiva, la agresivi-dad, la regulación del apetito y el apego. Se ha visto que en elSPW están disminuidas las neuronas secretoras de oxitocina enel núcleo paraventricular del hipotálamo. También se ha podidocomprobar que en el SPW existe un elevado nivel de oxitocinaen el líquido cefalorraquídeo. De ello se concluye que los nivelesalterados de oxitocina pueden relacionarse con falta de saciedad,compulsión, pellizcamiento de la piel y otras conductas dentrodel espectro de manifestaciones obsesivocompulsivas [7].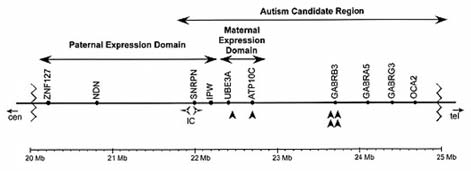 SÍNDROME X FRÁGIL
SÍNDROME X FRÁGIL