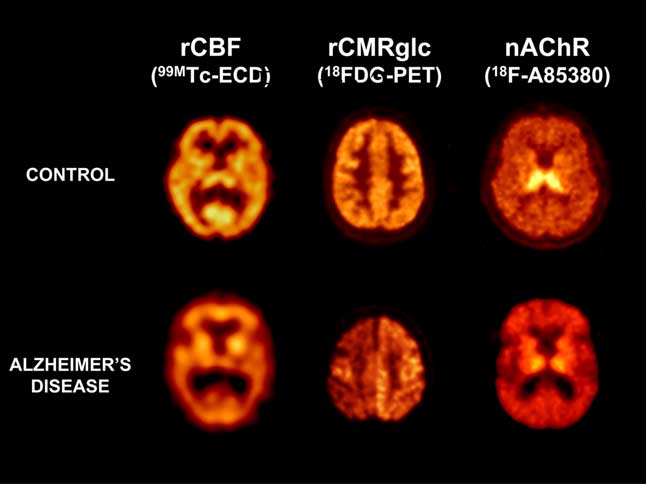
 Structural neuroimaging techniques, such as computed tomogra-
phy (CT) and magnetic resonance imaging (MRI), are routinely
used in the clinical evaluation of AD patients.
Structural neuroimaging techniques, such as computed tomogra-
phy (CT) and magnetic resonance imaging (MRI), are routinely
used in the clinical evaluation of AD patients.Microsoft word - publications_2010
Department of Clinical Physiology, Nuclear Medicine and PET Publications 2010 Doctoral theses and PhD theses defended during the year of 2010 De Nijs, R. Corrections in clinical Magnetic Resonance Spectroscopy and SPECT: Motion correction in MR spectroscopy, Downscatter correction in SPECT. Defended March 2nd 2010 at Technical University of Denmark, Department of Informatics and Mathematica